|
John writes ...  20-week foetus. Credit: Steve Allen Photography via Alamy. Introduction. Sickle-cell anaemia is a condition that occurs predominantly amongst people of Afro-Caribbean and Indian heritages. Eighty percent of cases occur in sub-Saharan Africa (see map) but we also note that, on a per capita basis, the Caribbean region has the second highest rate of occurrence (1). The name of the disease comes from the shape of the red blood cells which, instead of being round and squishy, are sickle-shaped and rather stiff. This means that they don’t travel so well through the smaller blood vessels which are thus prone to blockage, leading to a range of symptoms, including severe pain, as described in the NHS information page (2). Further, these aberrant red blood cells have a much shorter life in the body than normal red cells. This means that rates of cell production in the bone marrow may not keep up with the body’s needs, leading to anaemia. 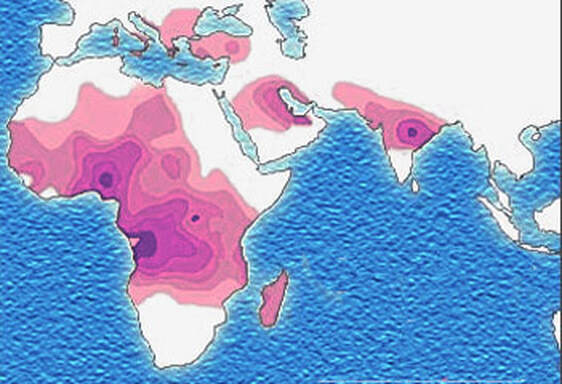 Distribution of sickle-cell disease in Africa and Eurasia. Credit: Muntuwandi at English Wikipedia, via GNU Free Documentation Licence. 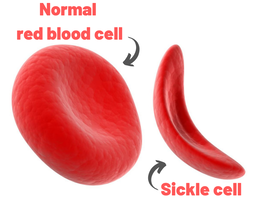 Normal and sickle-shaped red blood cells. Credit: public domain. Normal and sickle-shaped red blood cells. Credit: public domain. The sickle-shape of the red blood cells is caused by change in the structure of the oxygen-carrying protein, haemoglobin. Intriguingly, at normal oxygen concentrations, the oxygen-carrying capacity of the mutant haemoglobin is only slightly affected. However, it is when the protein ‘discharges’ its cargo of oxygen in the tissues where it is needed, that the change in its structure has its effect. The globin molecules stick together leading to the changes in the red cells that I mentioned earlier.  Fred Sanger. Credit: Public Domain Fred Sanger. Credit: Public Domain Some history. At this point we need to remind ourselves firstly that the building blocks of protein are called amino acids and secondly that the order of the building blocks (‘bases’) in DNA ‘tells’ the cell which amino acids to insert into a growing protein (Chapter 5 in the book). The order of bases in the DNA is thus the genetic code in which a group of three bases (a codon) corresponds to a particular amino acid and it is interesting that the biochemistry of haemoglobin has a significant role in our knowledge of this. Firstly, thanks mainly to the work of Fred Sanger in Cambridge, it was already possible in the late 1950s to work out the order of amino acids in protein. Sanger was awarded the 1958 Nobel Prize for Chemistry in recognition this work. Thus, the amino sequences of normal and sickle-cell haemoglobin could be compared and by 1959, Vernon Ingram, an American scientist working in Cambridge, had shown that the two differed by just one amino acid. In sickle-cell haemoglobin, a molecule of glutamic acid is replaced by a molecule of valine (3). Secondly, it was not long after, in 1961, that scientists started to crack the genetic code, a task that was completed by 1966. Marshall Nirenberg and Gobind Khorana were awarded the 1968 Nobel Prize for their major roles in this achievement. This meant that the presence of valine instead of glutamic acid in sickle-cell haemoglobin could be ascribed to a mutation in DNA in which the codon GAG is replaced the codon GTG (we recall that we often refer to the bases in DNA just by their initials, A, C, G, T). In other words, a mutation which involved the change of just one base was responsible for the aberrant behaviour of the mutant haemoglobin. GAG in DNA → Glutamic acid in protein GTG in DNA → Valine in protein This was the very first elucidation of a point mutation involving just one base which leads to the synthesis of a malfunctioning protein. It is worth reminding ourselves that this was achieved over a decade before Sanger (again!) published a method for sequencing DNA, a milestone which led to his receiving, in 1980, another Nobel Prize for Chemistry. Although other methods were developed at around the same time, the Sanger method was the first choice of most researchers, including me, from its arrival on the scene in 1977 until around 2010, when two much faster methods became widely adopted. In relation to sickle-cell disease, Sanger’s method was immediately used to sequence the normal and mutant globin genes, directly demonstrating the GAG to GTG change that had been deduced in previous research (as described above). Back to the womb. In the Bible, Nicodemus asked incredulously whether it was possible to return to his mother’s womb, with the strong implication that it was not (4). Of course, he was right. Here our figurative return to the womb involves looking at the haemoglobin circulating in the foetal blood stream. As in post-natal life, the haemoglobin has to pick up oxygen and deliver it round the body but from where does it get the oxygen? The only source available is the mother’s blood stream circulating through the placenta. The maternal and the foetal bloodstreams do not mix and so this oxygen capture occurs across cellular membranes. The transfer from the maternal to the foetal bloodstreams happens because foetal haemoglobin has a higher affinity (‘grabbing power’) for oxygen than maternal haemoglobin. It can therefore pull the oxygen from the mother’s bloodstream to the foetus’s bloodstream. If you find that hard to imagine, think of a magnet that has picked up some paper clips; if a stronger magnet is brought into the vicinity, it will ‘capture’ the paper clips from the weaker magnet (5). At birth, a remarkable genetic event starts to occur: the gene encoding foetal haemoglobin is down-regulated by the activity of a repressor gene (known as BCL11A) and the gene encoding adult haemoglobin is switched on. By the time an infant is about six months old, the red cells contain, almost exclusively, adult haemoglobin although most of us continue to make a tiny amount of foetal haemoglobin (about 1% of the total) throughout life. Furthermore, when we look at that small amount of persistent foetal haemoglobin it is clear that it does not carry the sickle-cell mutation, even in people with sickle-cell disease. Genome editing can cure sickle-cell disease. The gene-switching phenomenon that I describe above immediately raises the possibility of reversing the switch as a route to curing sickle-cell disease. Indeed, over the past two to three years there have been reports of the success of trials using exactly this approach. The story of one patient, Jimi Olaghere, is told in this BBC article: Sickle cell: ‘The revolutionary gene-editing treatment that gave me new life’ – BBC News.  Jimi Olaghere, cured of sickle-cell disease by the gene-editing procedure. Credit: Jimi Olaghere. In practice, the procedure is quite complex but the principle is clear. A patient’s bone marrow stem cells (from which blood cells are produced) are removed. Genome editing is used to inactivate both the mutant adult haemoglobin gene and the repressor that normally switches off the foetal haemoglobin gene. The bone marrow stem cells are put back in the patient who then starts producing foetal haemoglobin. We need to say at this point that because foetal haemoglobin is better at grabbing oxygen than adult haemoglobin it is also less good at releasing it out in the body’s tissues. This is not a problem for a foetus whose oxygen demands are not great but for a child or adult, this probably means that activities with high oxygen demand, including many sports, may be difficult (although golf is clearly possible). Nevertheless, it is obvious from the testimonies of patients like Jimi Olaghere that their lives are so much better after the treatment than before, including being able to play golf without the sickle-cell-associated pain. I started to compose this blog post on November 13th because I knew of the successful trials and I also learned that regulatory authorities in the USA, the EU and the UK were looking at the trials with a view to authorising the use of this gene-editing procedure in clinical practice. It was therefore very gratifying to turn on the BBC News on November 16th to hear that the UK’s Medicines and Healthcare Products Regulatory Agency (MHRA) had approved the procedure for treating both sickle-cell disease and thalassaemia (see below). This was also reported by several daily newspapers (6) and by New Scientist in its daily news (7). Thalassaemia. As mentioned above, the MHRA has also authorised the gene-editing procedure for use in treating another inherited haemoglobin disorder, thalassaemia, a move that was warmly welcomed by the Cyprus-based Thalassaemia International Federation (8). The name of the disease comes from the Greek word for sea and the name of the sea goddess, Thalassa (Θάλασσα) in Greek mythology. This is because people living around the eastern Mediterranean exhibit some of the highest incidences of thalassaemia; in an earlier publication (9) I describe programmes in Cyprus aimed at reducing the incidence of the disease. The seriousness of thalassaemia depends on which mutation or mutations a person has but for people with the most serious versions, life can be very difficult. I do not have the space here to describe the genetics or the symptoms in any more detail (see references for further information). However, I do want to emphasise that the trials of the gene-editing procedure described above also involved thalassaemia patients and were equally successful. A pause for thought. Overall response to this development has been very positive and rightly so: it is a brilliant use of excellent science. However, I want to pause for a moment to refer back to the opening paragraph. Eighty percent of cases of sickle-cell disease occur in sub-Saharan Africa with high incidences also seen in India and in the Caribbean region. The gene-editing-based treatment is very expensive, although in the UK, the USA and western Europe it is considered that, in cold accounting terms, the cost of this treatment is less than the lifetime costs of dealing with the medical needs of someone with sickle-cell disease or thalassaemia. That may be so but we still need to ask whether the treatment can be made available to the low and middle-income countries where the need is greatest.  David Liu. He and his research team at Harvard University were pioneers of DNA base-editing. Credit: Casey Atkins Photography, courtesy of the Broad Institute A look to the future. In my blog post for December 2022, I mentioned that a very accurate form of genome editing called DNA base-editing had been used to treat a teenage girl who had T-cell acute lymphoblastic leukaemia. Given that sickle-cell disease is caused by single-base mutation, would it be possible to use base-editing to change the sickle-cell haemoglobin gene back to the normal haemoglobin gene, i.e., to change GTG back to GAG? Well, the answer is not quite. It has not been possible to change the T back to A but it is possible to change it to a C, giving the codon GCG which codes for the amino acid alanine (10). Although this is clearly not the ‘original’ glutamic acid, haemoglobin carrying this change works more or less normally, so perhaps base-editing may eventually become the treatment of choice. John Bryant Topsham, Devon November 2023 (1) https://doi.org/10.1016/S2352-3026(23)00205-3
(2) Sickle cell disease - NHS (www.nhs.uk) (3) https://doi.org/10.1016/0006-3002(59)90183-0. (4) Holy Bible, John’s Gospel, Chapter 3, verse 4. (5) I am grateful to Dr Mark Bryant for this analogy. (6) For example, The Guardian UK medicines regulator approves gene therapy for two blood disorders | Gene editing | The Guardian. (7) Casgevy: Sickle cell CRISPR 'cure' is the start of a revolution in medicine | New Scientist. (8) TIF applauds new thalassaemia therapy | Cyprus Mail (cyprus-mail.com). (9) Bryant JA & La Velle L (2019) Introduction to Bioethics (2nd edition), Wiley, Chichester, pp. 122-123. (10) Gene editing shows promise as sickle cell therapy — Harvard Gazette.
0 Comments
|
AuthorsJohn Bryant and Graham Swinerd comment on biology, physics and faith. Archives
July 2024
Categories |
 RSS Feed
RSS Feed
